| disease | Congenital Esophageal Atresia |
Among various congenital esophageal anomalies, esophageal atresia is the most common, accounting for approximately 85%. About 20% of congenital esophageal atresia cases are accompanied by congenital cardiovascular anomalies, and another 10% are associated with anal atresia.
bubble_chart Pathological Changes
Congenital esophageal atresia can be classified into five types based on the location of the atresia and the presence or absence of tracheoesophageal fistula (Figure 1).
|
|
|
|
|
| Type I 86.5% | Type II 0.8% | Type III 7.7% | Type IV 0.7% | Type V 4.2% |
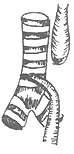
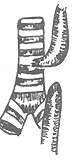
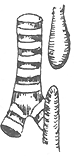
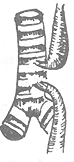
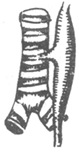
Figure 1 Types of congenital esophageal atresia
1. The proximal esophagus ends blindly, while the distal segment connects to the posterior wall of the trachea, forming a tracheoesophageal fistula. According to Holder's statistics, this is the most common type, accounting for approximately 86.5%.
2. The proximal esophagus connects to the posterior wall of the trachea, forming a tracheoesophageal fistula, while the distal segment ends blindly. This type is rare, accounting for about 0.8%.
3. Both the proximal and distal segments of the esophagus end blindly and do not connect to the trachea, with no tracheoesophageal fistula. This type accounts for 7.7%.
4. Both the proximal and distal segments of the esophagus separately connect to the posterior wall of the trachea, forming two tracheoesophageal fistulas. This type is also very rare, accounting for 0.7%.
5. The esophageal lumen is patent without atresia, but the anterior wall of the esophagus connects to the posterior wall of the trachea, forming a tracheoesophageal fistula, accounting for 4.2%.
bubble_chart Clinical Manifestations
The typical clinical manifestations of esophageal atresia include the inability to swallow saliva, which refluxes into the mouth, leading to drooling and foaming at the mouth immediately after birth. During each feeding, in type 1 and type 3 patients, milk cannot pass into the stomach and instead spills into the respiratory tract. In type 2, type 4, and type 5 cases, milk directly enters the trachea, causing choking, vomiting, respiratory distress, cyanosis, and a high risk of aspiration pneumonia. In type 1 and type 4 cases with a tracheoesophageal fistula between the lower esophagus and trachea, air from the respiratory tract can enter the gastrointestinal tract through the fistula, leading to abdominal distension and fullness. At the same time, gastric fluid can reflux into the respiratory tract via the tracheoesophageal fistula, causing aspiration pneumonia, fever, and shortness of breath. Since food cannot enter the gastrointestinal tract, the infant becomes dehydrated and emaciated. Without timely treatment, death may occur within days due to pulmonary inflammation and severe dehydration. Physical examination often reveals signs of dehydration and excessive saliva in the mouth. In cases complicated by pneumonia, rales may be heard in the lungs, and the affected areas may exhibit dullness on percussion.
bubble_chart DiagnosisA thin, soft catheter is inserted through the mouth or nose and becomes obstructed at the upper esophageal blind pouch, unable to pass into the stomach. Injecting 0.5–1.0 ml of water-soluble iodine contrast agent through the catheter can reveal the position and length of the upper esophageal blind pouch and determine whether there is a fistula between the upper esophagus and the trachea. Abdominal X-ray findings of gas in the gastrointestinal tract indicate that the lower esophagus communicates with the trachea, confirming the presence of a tracheoesophageal fistula. For Type 5 patients with no esophageal atresia but with a tracheoesophageal fistula, although they may not exhibit symptoms like drooling or inability to eat, ingestion—especially of liquid foods—can cause choking if some food enters the trachea through the fistula. In cases with a smaller fistula, choking after eating may occur intermittently. Some patients may not show symptoms until several years after birth. Diagnosis of this type requires confirmation through esophagoscopy.
bubble_chart Treatment Measures
Congenital esophageal atresia cases die within a few days after birth without treatment, so surgery should be performed as early as possible after a clear diagnosis to correct the deformity. Preoperative care should include appropriate fluid replacement to correct dehydration and electrolyte imbalances; prevention and treatment of aspiration pneumonia; administration of antibiotics; and maintaining normal body temperature. Placing the infant in a semi-sitting position can reduce the risk of gastric fluid flowing into the respiratory tract. Insert a thin catheter into the upper esophageal lumen for continuous negative pressure suction, or frequently remove oral secretions to prevent or reduce the inhalation of oral secretions into the lungs. For cases with lung inflammation or atelectasis, ventilation function should be maintained, and oxygen inhalation should be provided.
**Surgical Procedure:** A right posterolateral thoracotomy is typically used. The 4th or 5th rib is resected to enter the chest through the rib bed or via the 4th intercostal space. The azygos vein is ligated and divided either extrapleurally or after opening the pleura. The lower esophageal segment is mobilized, and a thin tape is placed around it for traction to expose the tracheoesophageal fistula on the posterior tracheal wall. The fistula is transected approximately 3 mm from the tracheal wall, and the tracheal incision is closed with 3–4 interrupted transverse sutures using 5–0 sutures, then covered with adjacent pleura. Preserving a short segment of the fistula tissue prevents stenosis at the tracheoesophageal fistula suture site. When mobilizing the lower esophageal segment, the operation should be gentle, and the mobilized length should not be excessive to avoid compromising blood supply. The upper esophageal segment has a richer blood supply and should be sufficiently mobilized to achieve adequate length for anastomosis with the lower segment and to reduce anastomotic tension. Preoperative placement of a catheter in the upper esophagus aids in identification and mobilization. After confirming that the lengths of the upper and lower esophageal segments are sufficient for end-to-end anastomosis, the apex of the lower esophageal blind pouch is excised to expose the lumen. The muscular layer of the upper esophageal wall is bluntly dissected upward by 6–8 mm at the distal end. The mucosal layer of the upper esophageal wall is then anastomosed end-to-end with the full thickness of the lower esophageal wall. Two traction sutures are placed at each end of the anastomosis, followed by interrupted sutures of the posterior and anterior walls. The muscular layer of the upper esophageal wall is then pulled down and sutured over the anastomosis to cover it (Figure 1).
(1) **Positioning and 4th Intercostal Incision**
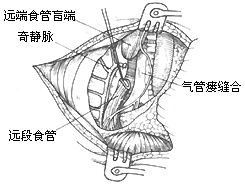
(2) **Division and Suture of Tracheoesophageal Fistula**
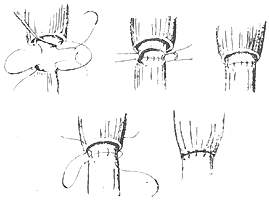
(3) **End-to-End Esophageal Anastomosis** The mucosal layer of the upper esophageal wall is sutured to the full thickness of the lower esophageal wall with interrupted sutures, and the muscular layer of the upper esophageal wall is sutured over the anastomosis.
**Figure 1** Initial stage [first stage] corrective surgery for congenital esophageal atresia
(3) **End-to-End Esophageal Anastomosis**
The mucosal layer of the upper esophageal wall is sutured to the full thickness of the lower esophageal wall with interrupted sutures, and the muscular layer of the upper esophageal wall is sutured over the anastomosis.
Alternatively, the muscular layer of the upper esophageal wall may not be dissected, and the anastomosis can be performed using the full thickness of both the upper and lower esophageal walls. Before completing the anterior wall sutures, a thin catheter pre-inserted nasally or orally is passed through the anastomosis into the stomach for postoperative decompression and feeding. Alternatively, a gastrostomy can be performed for postoperative feeding. A drainage tube is placed extrapleurally or intrapleurally and removed about one week postoperatively after X-ray confirmation of anastomotic healing. Oral feeding can then begin. Esophageal anastomotic dilation can be started three weeks postoperatively to achieve an internal diameter of at least F24.
For premature infants with a weight deficit of 2kg, generally in poor condition or with other severe congenital organ malformations, staged corrective surgery is required. During the initial stage [first stage] of the operation, the esophageal-tracheal fistula is severed and sutured, and a gastrostomy is performed through an abdominal incision to provide nutrition. A catheter is placed in the upper esophagus for continuous negative pressure suction or a cervical esophagostomy is performed to prevent aspiration pneumonia. Several weeks later, once the weight increases to approximately 3kg, the intermediate stage [second stage] surgery is performed to anastomose the upper and lower segments of the esophagus end-to-end.
In patients where both the upper and lower segments of the esophagus are blind ends without a tracheoesophageal fistula, the length of the esophagus is often insufficient for end-to-end anastomosis. This type of congenital esophageal atresia also requires staged surgery. The initial stage [first stage] involves bringing the proximal esophageal segment out through a neck incision, opening the blind end of the proximal esophagus to allow saliva drainage and prevent aspiration into the respiratory tract. Additionally, a gastrostomy is performed through an abdominal incision for feeding. When the child grows to 3–4 years old, the intermediate stage [second stage] of colonic interposition is performed to replace the esophagus.


