| disease | Chronic Cardiac Insufficiency |
| alias | Congestive Heart Failure, Pump Failure |
Cardiac insufficiency is defined as a clinical syndrome characterized by impaired cardiac systolic and diastolic function caused by various disease etiologies. It progresses to a stage where cardiac output cannot meet the metabolic demands of systemic blood flow despite normal circulating blood volume and vasomotor function, leading to hemodynamic abnormalities and activation of the neurohormonal system. This condition is also referred to as cardiac insufficiency syndrome or heart failure syndrome. Originally, "pump failure" specifically denoted left heart failure in acute myocardial infarction. However, it is now broadly used to describe impaired cardiac pumping function caused by different disease etiologies. Traditional views held that all patients with cardiac insufficiency exhibited symptoms of organ congestion, hence the term "congestive heart failure." A newer perspective categorizes cardiac insufficiency into asymptomatic and symptomatic stages. The asymptomatic stage presents objective evidence of ventricular dysfunction (e.g., reduced left ventricular ejection fraction) without typical congestive heart failure symptoms, corresponding to NYHA (New York Heart Association) Class I. This stage is considered pre-symptomatic heart failure and, if left untreated, will eventually progress to symptomatic cardiac insufficiency. Clinically, cardiac insufficiency can manifest as acute, chronic, or compensated forms, depending on the speed of onset and the degree of circulatory system compensation. Advances in ventricular diastolic function assessment have further enabled differentiation between systolic dysfunction-predominant and diastolic dysfunction-predominant cardiac insufficiency. Chronic primary myocardial diseases or long-term ventricular pressure/volume overload can lead to primary or secondary impairment of myocardial contractility. In the early stages, compensatory mechanisms maintain stroke volume and cardiac output to meet metabolic demands at rest and during activity. However, in late-stage (Stage III), even maximal compensation fails to sustain adequate stroke volume and cardiac output. The former is termed the compensated phase of chronic cardiac insufficiency (also called latent, compensated, or asymptomatic cardiac insufficiency), while the latter is the decompensated phase (or decompensated cardiac insufficiency). Since decompensated chronic cardiac insufficiency often involves organ congestion (or static blood), it is commonly termed congestive heart failure or symptomatic heart failure.
bubble_chart EtiologyThe most common disease causes of adult congestive heart failure are coronary atherosclerotic heart disease (coronary heart disease), hypertensive heart disease (dilated cardiomyopathy), valvular disease, cardiomyopathy, and pulmonary heart disease. Other relatively common disease causes include myocarditis, nephritis, and congenital heart disease. Less common and easily overlooked disease causes include pericardial diseases, hyperthyroidism and hypothyroidism, anemia, beriberi, arteriovenous fistula, atrial myxoma and other heart tumors, connective tissue diseases, high-altitude sickness, and rare endocrine disorders.
The aforementioned fundamental causes of heart failure can affect cardiac function through the following mechanisms, leading to heart failure.
(1) Primary impairment of myocardial contractility: This includes myocardial infarction, myocardial inflammation, degeneration, or necrosis (such as rheumatic or viral myocarditis, diphtheritic myocardial necrosis), myocardial hypoxia or fibrosis (such as coronary heart disease, pulmonary heart disease, cardiomyopathy, etc.), as well as metabolic or toxic changes in the myocardium, all of which weaken myocardial contractility and lead to heart failure.
(2) Excessive pressure load (afterload) on the ventricles: Pulmonary and systemic hypertension, left and right ventricular outflow tract stenosis, and aortic or pulmonary valve stenosis can all increase resistance during ventricular contraction, exacerbating afterload and leading to secondary impairment of myocardial relaxation and contraction, resulting in heart failure.
(3) Excessive volume load (preload) on the ventricles: Valvular insufficiency, intracardiac or large vessel left-to-right shunts, etc., increase ventricular diastolic volume and exacerbate preload, which can also lead to secondary weakening of myocardial contractility and heart failure.
(4) Hyperkinetic circulatory state: This mainly occurs in conditions such as anemia, systemic arteriovenous fistula, hyperthyroidism, and beriberi heart disease. Due to reduced peripheral vascular resistance and increased cardiac output, ventricular volume load is exacerbated, leading to heart failure.(5) Insufficient ventricular preload: Conditions such as mitral stenosis, cardiac tamponade, and restrictive cardiomyopathy restrict ventricular filling and cause congestion in the systemic and pulmonary circulation.
Precipitating factors of heart failure: Analysis of domestic clinical data shows that 89.8% of heart failure episodes have precipitating factors. Common precipitating factors are as follows:
1. Infection: Respiratory infections are the most common, followed by rheumatic fever. In children, rheumatic fever ranks first. Urinary tract infections are also common in female patients. Subacute infective endocarditis often damages heart valves and myocardium, precipitating heart failure.
2. Excessive physical activity and emotional stress.
3. Excessive sodium intake.
4. Arrhythmias: Particularly rapid arrhythmias, such as atrial fibrillation (AF) or atrial flutter (AFL) with rapid ventricular rates.
5. Pregnancy and childbirth.
6. Excessive or rapid intravenous infusion (especially sodium-containing fluids) or blood transfusion.
7. Overdose or deficiency of digitalis.
8. Drug effects: ① Use of drugs that inhibit myocardial contractility, such as beta-blockers, drugs that deplete catecholamines (e.g., reserpine-like drugs), sympathetic ganglion blockers (e.g., guanethidine), and certain antiarrhythmic drugs (e.g., quinidine, procainamide, verapamil, etc.); ② Water and sodium retention caused by hormones and drugs, such as adrenal corticosteroids.
9. Others: Hemorrhage and anemia, pulmonary embolism, ventricular aneurysm, uncoordinated myocardial contraction, papillary muscle dysfunction, etc.bubble_chart Pathological Changes
Pathological Anatomy: The pathological changes in congestive heart failure include: ① Compensatory changes in the heart itself, such as myocardial hypertrophy and cardiac chamber dilation; ② Pathological changes caused by long-term increased venous pressure, leading to organ congestion; ③ Mural thrombi in the atria and ventricles, venous thrombosis, {|###|} embolism, and organ infarction. In patients with long-term left ventricular or left atrial failure, pulmonary capillary congestion occurs, with thickening of the pulmonary {|###|} and middle layer of pulmonary veins, and varying degrees of fibrosis in the inner membrane. There is an increase in hemosiderin-laden macrophages in the alveolar spaces, thickening of alveolar walls, and reduced elasticity. In patients with long-term right heart failure, capillary and venous {|###|} stasis occurs in visceral organs, with central hepatic sinusoid stasis, which in severe cases can lead to central lobular hepatocyte necrosis and connective tissue proliferation, resulting in cardiac cirrhosis. Mural thrombi within the cardiac chambers are a relatively specific pathological change in heart failure, commonly found in the left and right atrial appendages and the apex of the left ventricle. Venous thrombosis is mostly caused by sluggish blood flow, frequently occurring in the lower limb veins, and the proximal end of the thrombus is prone to breakage, potentially causing pulmonary embolism and varying degrees of pulmonary infarction. Detachment of mural thrombi from the left side of the heart can lead to systemic {|###|} embolism, often occurring at branches of the abdominal aorta or aortic bifurcation, causing infarction in the brain, kidneys, limbs, spleen, and mesentery. Detachment of mural thrombi from the right side of the heart causing pulmonary embolism is less common.
Long-term increased load leads to myocardial hypertrophy and cardiac chamber dilation, culminating in heart failure. The corresponding pathological changes can be roughly divided into three stages:
**Initial Stage [First Stage]**: A brief injury period. Clinically, pulmonary congestion is observed, and pathological examination reveals myocardial fiber edema and widened separation. Intracellular glycogen and adenosine triphosphate (ATP) levels decrease, phosphocreatine significantly reduces, lactate production slightly increases, protein synthesis is vigorous, and ribonucleic acid (RNA) and mitochondria increase.
**Intermediate Stage [Second Stage]**: A relatively long-term, stable period of hyperfunction. Clinical symptoms are not obvious, and histopathology shows myocardial hypertrophy, enlarged myocardial fibers, and minor fibrotic lesions. Intracellular glycogen, ATP, and phosphocreatine levels are normal, lactate production increases, protein synthesis is normal, RNA content is normal, and deoxyribonucleic acid (DNA) decreases. The increase in myocardial fibers is relatively more pronounced than the increase in mitochondria.
**Late Stage [Third Stage]**: A prolonged period of exhaustion and fibrosis. Clinically, heart failure persists, and histopathology reveals replacement of myocardial tissue by fibrous tissue, disproportionate connective tissue proliferation, fatty degeneration, and pyknosis of muscle cell nuclei. Intracellular protein synthesis and DNA are significantly reduced, with other features similar to the intermediate stage [second stage].
Microscopically, there is no direct evidence of morphological abnormalities in contractile proteins.
**Pathophysiology**:
(1) **Normal Cardiac Pumping Physiology**: Although the pumping function of the heart primarily depends on myocardial contraction and relaxation characteristics, it is also influenced by preload, afterload, and heart rate.
1. Characteristics of Myocardial Contraction and Relaxation The sarcomere is the fundamental unit of myocardial contraction and relaxation, composed of interdigitated thick and thin filaments. The thick filaments are myosin, located at the center of the sarcomere. The thin filaments are actin, positioned on either side of the sarcomere and partially overlapping with myosin. During myocardial relaxation, the complex of two regulatory proteins on actin—troponin and tropomyosin—prevents its binding to myosin, keeping the two separated and the sarcomere extended. When myocardial cells depolarize, calcium ions outside the membrane flow inward alongside sodium ions, entering the sarcotubular system (including the sarcoplasmic reticulum and transverse tubule system) through the membrane. A small influx of Ca2+ stimulates the release of stored calcium ions from the sarcoplasmic reticulum via the Ca2+ release channel RYR receptor. The Ca2+ acts on the regulatory protein complex on actin, exposing the binding sites on actin and allowing the globular heads of myosin to bind to actin, forming cross-bridges. Actin filaments slide toward the center of the sarcomere, causing the sarcomere to shorten and the myocardium to contract. The process by which myocardial cell depolarization leads to mechanical contraction is called excitation-contraction coupling. The energy required for myocardial contraction is provided by ATP, produced by mitochondria through the action of actomyosin ATPase. The strength and speed of myocardial contraction depend on sarcomere length (normally 2.0–2.2 μm) and, more critically, on calcium ion transport and energy supply. Stimulation of the β1 receptors located on the outer layer of the muscle fiber membrane activates the associated guanine nucleotide regulatory protein Gs. This, in turn, activates adenylate cyclase, converting ATP to cAMP, which activates protein kinase, leading to phosphorylation of calcium channels. This increases calcium ion influx and rapid uptake by the sarcoplasmic reticulum, enhancing excitation-contraction coupling and thereby increasing myocardial contractility. Inhibition of phosphodiesterase can prevent cAMP degradation, also raising intracellular calcium ion concentration and augmenting myocardial contractility.
After excitation-contraction coupling, the sarcoplasmic reticulum reuptakes Ca2+, the sarcolemma sodium-calcium exchange and the sarcolemma Ca2+ pump transport Ca2+ outside the sarcolemma, the intracellular Ca2+ concentration decreases, the regulatory protein complex separates from calcium ions, and the regulatory protein acts on the binding sites of actin, causing the cross-bridges between contractile proteins to detach. Actin slides back to its original position on both sides, the sarcomere relaxes, and the myocardium dilates. The energy consumed during myocardial diastole is greater than during contraction. When energy supply is insufficient, such as in myocardial ischemia or ventricular wall hypertrophy, diastolic function is impaired earlier than systolic function.
2. Preload refers to the volume load that the ventricle bears before contraction, i.e., the end-diastolic volume of the ventricle. It is influenced by circulating blood volume, venous tension, ventricular compliance, and atrial contraction. According to the Frank-Starling law, an increase in preload, by stretching the muscle fibers, enhances myocardial contractility and stroke volume within certain limits. However, when preload exceeds a certain threshold, excessive stretching of the muscle fibers (>2.2 μm) leads to a decrease in myocardial contractility and stroke volume. Although recent electron microscopy studies show that the average sarcomere length in failing hearts in vivo is around 2.2 μm, not operating on the descending limb of the Frank-Starling curve, this suggests that the decline in myocardial contractility is primarily due to intrinsic defects (see below). Clinically, ventricular end-diastolic pressure (i.e., filling pressure) is often used to represent ventricular preload, and the ventricular function curve (Figure 1) illustrates the relationship between preload and stroke volume. For the left ventricle, stroke volume peaks when end-diastolic pressure is between 2.0–2.4 kPa (15–18 mmHg). Insufficient or excessive preload can both lead to reduced stroke volume. In cardiac insufficiency, the ventricular function curve shifts downward and to the right, and the increase in stroke volume with preload is significantly diminished.
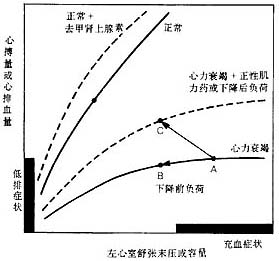
Figure 1 Left Ventricular Systolic Function Curve
In a normal left ventricle, stroke volume or cardiac output increases with preload (left ventricular end-diastolic pressure or volume) until reserves are exhausted. In heart failure, the left ventricular function curve is depressed and shifts downward and to the right. Using positive inotropic agents or reducing afterload can enhance myocardial contractility, allowing the left ventricle to maintain cardiac output at higher end-diastolic pressures or volumes. Reducing preload (e.g., with diuretics or venous dilators) can move point A to point B, alleviating congestion symptoms, but cardiac output does not increase and may even decrease. Using positive inotropic agents or reducing afterload can move point A to point C, improving both congestion and low-output symptoms.
3. Afterload refers to the pressure load the ventricle bears during ejection, including wall tension and vascular resistance. According to Laplace's law, wall tension is directly proportional to intraventricular pressure and chamber radius and inversely proportional to wall thickness. Vascular resistance primarily depends on peripheral vascular resistance and is also influenced by aortic pressure, aortic wall compliance, intravascular blood volume, and blood viscosity. When afterload increases, the rate and extent of ventricular muscle contraction decrease, leading to reduced stroke volume. The effect of afterload on reducing stroke volume is more pronounced in cardiac insufficiency.
4. Heart rate directly affects cardiac output. An increase in heart rate within certain limits can correspondingly enhance myocardial contractility; however, when the heart rate is too fast, ventricular filling decreases due to a significantly shortened diastolic period, resulting in a decline in stroke volume and cardiac output.
Under normal circumstances, the body can regulate myocardial contraction and relaxation, as well as cardiac preload, afterload, and heart rate through the neurohormonal system and the heart-blood vessel system itself. This adjusts stroke volume to meet changes in the body's metabolic demands. For example, a normal heart at rest has a cardiac output of about 4L/min, while during intense exercise, sympathetic stimulation enhances myocardial contraction, accelerates heart rate, and selectively constricts blood vessels to redistribute blood flow. This can increase cardiac output up to 38L/min, with 70–80% directed to the working skeletal muscles. This demonstrates that when cardiac function is normal, the body's sophisticated regulatory systems can promptly coordinate the relationship between cardiac work and metabolic demands. In other words, a healthy heart possesses considerable reserve capacity to adapt to the body's metabolic needs.
(II) Changes in Cardiac Insufficiency When various causes lead to myocardial overload or loss of myocardium, the immediate and short-term regulation of circulatory function relies on the hemodynamic effects of the neurohormonal system, while long-term regulation depends on myocardial remodeling and ventricular remodeling induced by mechanical load and mediated by the neurohormonal system. If the body can maintain normal cardiac output at rest and during exercise through the above regulation, or normal at rest but slightly insufficient during exercise, it is considered good adaptation (adaptation), with compensatory heart pump function but reduced cardiac reserve. If left ventricular diastolic and systolic dysfunction still occurs despite the above regulation, it is considered maladaptation. If there is only objective evidence of left ventricular diastolic and systolic dysfunction without obvious subjective symptoms, it is called asymptomatic left ventricular dysfunction or asymptomatic heart failure. When left ventricular dysfunction progresses to the point where cardiac output is insufficient to meet systemic metabolic needs, and manifestations of heart failure such as low cardiac output, elevated left ventricular filling pressure, pulmonary and systemic congestion, and water-sodium retention occur, it is called symptomatic heart failure. In the symptomatic heart failure stage, activation of the neurohormonal system increases peripheral vascular resistance, afterload, and systolic wall stress; it also causes vasoconstriction of capacitance vessels, water-sodium retention, increased preload and diastolic wall stress, and further reduction in cardiac output. This vicious cycle leads to persistent and progressive worsening of heart failure. Systolic cardiac insufficiency is characterized by a decrease in ventricular ejection fraction; in diastolic cardiac insufficiency, ventricular ejection fraction is normal but ventricular filling is impaired. The latter also causes pulmonary congestion and reduced cardiac output, clinically manifesting as dyspnea and fatigue (lack of strength), which is difficult to distinguish from the former. Some patients may have both conditions simultaneously.
1. Myocardial Remodeling Triggered by mechanical signals such as increased ventricular wall stress, chemical signals such as adrenergic α1 or β receptor stimulation and angiotensin II AT1 receptor stimulation, as well as various peptide growth factors. These signals are transmitted through the sarcolemma channels and microtubule system (e.g., cAMP) and, upon reaching the DNA in the cell nucleus, induce changes in gene expression. Consequently, protein synthesis in myocardial cells accelerates, collagen synthesis exceeds degradation, myocardial cells hypertrophy, fibroblasts proliferate, and smooth muscle in myocardial microvessels proliferates, leading to thickening of the middle layer. The result is myocardial hypertrophy, changes in protein structural components, alterations in myocardial excitation-contraction coupling, and corresponding changes in generation and transformation responses and function (Table 6). These changes have both beneficial and adverse aspects.
Table 6: Changes in Myocardial Remodeling and Their Beneficial and Adverse Aspects
| Changes in gene expression of sarcolemma proteins and contractile proteins ↓ | |
| Prolonged action potential duration | Increased afterdepolarization |
| Increased Ca2+ transient amplitude | Increased triggered automaticity |
| Increased arrhythmias due to triggered mechanisms | |
| Isoproterenol stimulated sensitivity↓ | Heart rate variability↓ |
| Myocardial shortening rate↓ | Cardiac output↓ |
| Myocardial relaxation rate↓ | Ventricular filling volume↓ |
↓ | ↓ |
| Energy utilization | Arrhythmia Sudden death |
| Effect improvement | Heart failure Death |
In addition, the remodeling of non-myocyte components during myocardial remodeling can affect myocardial stiffness. Changes in coronary microvascular perivascular fibrosis also impair myocardial blood supply and reduce coronary reserve.
2. Ventricular remodeling Ventricular remodeling includes not only changes in myocardial hypertrophy and remodeling but also alterations in ventricular wall thickness, composition, chamber volume, shape, myocardial stiffness, and the structure of intramyocardial coronary arteries.
After transmural acute myocardial infarction, the left ventricle undergoes complex geometric and wall structure changes, including infarct expansion, hypertrophy of non-infarcted myocardium, longitudinal slippage of myocardial cells along the sides, and stretching of myocardial fibers. The myocardial cells are arranged in a circular pattern, increasing the chamber radius and transforming from the normal elliptical shape to a spherical one, with increased chamber compliance. This remodeling process can last for years. Persistent ventricular remodeling can lead to progressive chamber enlargement and heart failure.
Under pressure overload, hypertrophied myocardium exhibits increased transverse sarcomeres, enlarged cell diameter, and thickened ventricular walls with unchanged or reduced chamber volume, resulting in concentric hypertrophy. The growth of non-myocyte components is disproportionate to myocardial cell growth, with excessive collagen accumulation (increased collagen-to-myocardium volume ratio), interstitial or intramyocardial microvascular perivascular fibrosis, and thickening of the medial layer of resistance coronary arteries.
Under volume overload, hypertrophied myocardium exhibits increased longitudinal sarcomeres, elongated cells, relatively thinned ventricular walls, proportional growth of collagen and myocardial cells, or increased collagen degradation, leading to increased chamber compliance and volume, resulting in eccentric hypertrophy.
Initially, ventricular wall hypertrophy helps correct elevated systolic and diastolic wall stress, restoring it to normal, but some degree of elevated wall stress persists. Initially, chamber enlargement helps adjust the reduced cardiac output, increasing it somewhat, but diastolic wall stress rises. Persistent ventricular remodeling leads to progressive deterioration of ventricular systolic and diastolic function due to generation and transformation reactions and functional abnormalities caused by hypertrophic myocardial remodeling, increased myocardial stiffness, and altered chamber compliance (Figure 2). Additionally, reduced coronary reserve exacerbates the imbalance between energy supply and demand caused by myocardial remodeling, and the reduction in myocardial cell numbers increases the load on the remaining myocardium. This vicious cycle promotes the onset and progression of heart failure.
|
|
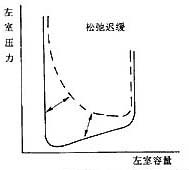
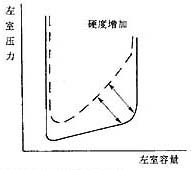
Figure 2 Changes in pressure-volume relationship during left ventricular diastolic dysfunction
The left panel shows delayed left ventricular relaxation, occurring in early diastole; the right panel shows increased left ventricular stiffness, demonstrating an advanced stage of diastole with increased dp/dv. Ventricular volume decreases.
3. Changes in the neurohormonal system Recent extensive research has shown that the activation of the neurohormonal system plays a role throughout the occurrence and progression of heart failure. Initially, neurohormonal activation may temporarily maintain circulation and perfusion of vital organs, but prolonged activation promotes myocardial remodeling and ventricular remodeling, ultimately leading to heart failure. At this stage, the activation of the neurohormonal system increases ventricular preload and afterload, further exacerbating hemodynamic disturbances.
(1) Activation of the sympathetic-adrenal system: A decrease in stroke volume or hypotension activates the sympathetic-adrenal system through baroreceptor-mediated reflexes. This increases sympathetic nerve impulses and the secretion of adrenal catecholamines, leading to the following changes: ① Increased heart rate; ② Myocardial β-receptor stimulation activates cAMP enzymes, raising intracellular cAMP levels and enhancing myocardial contractility; ③ Systemic vasoconstriction. Venous constriction increases venous return, boosting stroke volume via the Frank-Starling mechanism, while selective arteriolar constriction helps maintain blood pressure and ensure blood supply to vital organs; ④ Reduced renal perfusion pressure due to increased renal sympathetic activity stimulates renin release, activating the renin-angiotensin system; ⑤ Stimulation of α 1 and β receptors promotes myocardial growth. The degree of plasma norepinephrine (NE) elevation reflects the extent of sympathetic-adrenal system activation. Plasma NE levels are already significantly elevated in asymptomatic left ventricular systolic dysfunction and increase further as heart failure progresses, becoming markedly elevated in symptomatic stages. Patients with significantly elevated plasma NE levels generally have a poorer prognosis. Effective treatment can reduce markedly elevated plasma NE levels.
(2) Activation of the renin-angiotensin-aldosterone system: A decrease in stroke volume or hypotension reduces glomerular afferent arteriole pressure, while increased sympathetic activity stimulates juxtaglomerular cells to release renin. Renin hydrolyzes angiotensinogen (synthesized in the liver) to produce angiotensin I, which is converted to angiotensin II (Ang II) by angiotensin-converting enzyme (ACE), primarily located on pulmonary microvascular endothelial cells. Ang II binds to its receptors, producing the following physiological effects: ① Potent vasoconstriction; ② Positive inotropic effects on the myocardium; ③ Promotion of cardiomyocyte, cardiac fibroblast, and vascular smooth muscle cell growth; ④ Stimulation of norepinephrine release from sympathetic nerve terminals; ⑤ Promotion of aldosterone and vasopressin secretion; ⑥ Stimulation of adrenal deoxycorticosterone production; ⑦ Promotion of bradykinin degradation; ⑧ Inhibition of renin secretion. Aldosterone regulates potassium-hydrogen and potassium-sodium exchange in the distal renal tubules, leading to sodium retention and potassium excretion, along with water retention. Additionally, it promotes myocardial collagen fiber proliferation. Plasma renin activity, Ang II, and aldosterone levels reflect the degree of renin-angiotensin-aldosterone system activation. In asymptomatic left ventricular systolic dysfunction, plasma renin activity may be normal or mildly elevated (grade I), increasing progressively with heart failure progression and becoming significantly elevated in symptomatic stages. Effective treatment can reduce markedly elevated plasma renin activity.
(3) Increased vasopressin: When stroke volume decreases or hypotension severely affects tissue perfusion, vasopressin secretion increases. The antidiuretic and peripheral vasoconstrictive effects of vasopressin lead to water and sodium retention and increased ventricular afterload. In patients with asymptomatic left ventricular systolic dysfunction, plasma vasopressin levels may not increase or may show grade I elevation. There is considerable individual variation in whether plasma vasopressin levels increase in symptomatic heart failure patients.
(4) Increase in Atrial Natriuretic Peptide: Atrial natriuretic peptide (ANP), also known as atrial natriuretic factor (ANF), is primarily synthesized and secreted by atrial myocytes. The main mechanism triggering its release is increased atrial pressure or atrial stretch. Recently, ventricular natriuretic peptide (also known as brain natriuretic peptide) has been discovered. ANP has potent vasodilatory and natriuretic-diuretic effects, which help modulate the body's response to vasoconstrictive and sodium-retaining hormones. In the asymptomatic left ventricular dysfunction stage, ANP plays a dominant role in counteracting the vasoconstriction and increased intravascular volume caused by activation of the sympathetic-adrenal system. However, as ventricular function continues to deteriorate, although plasma ANP levels rise further, its compensatory effects are ultimately offset and overwhelmed by the vasoconstrictive and sodium-retaining actions of the aforementioned three neurohormones.
(5) Changes in Local Tissue Hormone Systems: Recent studies have confirmed that tissues such as the heart, blood vessels, and kidneys can independently produce and secrete hormones that act on themselves or neighboring cells, separate from the systemic endocrine system. This is referred to as tissue autocrine and paracrine systems. Within the myocardium (including cardiomyocytes, interstitial cells, and microvascular cells), vasoconstrictive, positive inotropic, and growth-promoting hormones (e.g., Ang II, endothelin) are produced, acting on local myocardial microvessels and cardiomyocytes. At the same time, opposing hormones are also produced, such as vasodilatory microvascular, negative inotropic, and growth-inhibiting hormones (e.g., endothelium-derived relaxing factor, ANP, prostaglandin E2, and prostaglandin I2). These systems maintain a balanced regulatory relationship. When myocardial overload or loss occurs, the myocardial autocrine and paracrine systems are activated, with vasoconstrictive, positive inotropic, and growth-promoting hormones becoming dominant. Evidence suggests that even when the circulating neuroendocrine system tends to return to normal or remains at a low activation level, the myocardial autocrine and paracrine systems remain persistently activated, mediating ongoing myocardial remodeling and ventricular remodeling, which can persist for an extended period.
Recent studies have also identified the presence of human myocardial chymase in the heart, which is increased in failing myocardium compared to normal. Most of the conversion of Ang I to Ang II in the myocardium occurs through this enzyme, with only a small portion mediated by ACE. ACE inhibitors cannot inhibit the action of this enzyme.
4. Changes in Peripheral Vasculature: The aforementioned neurohormonal regulation leads to constriction of peripheral arteries and veins. Contraction of peripheral small stirred pulses increases peripheral resistance. The responsiveness of baroreceptor regulation is weakened, so the use of vasodilators often does not elicit a significant heart rate increase. To ensure adequate blood supply to vital organs such as the brain and heart despite limited cardiac output, blood circulation to the skin, viscera, skeletal muscles, and kidneys is often significantly reduced. This regional redistribution of blood flow also contributes to some of the symptoms and signs of heart failure.
During the pathophysiological changes of heart failure, the overshoot of compensatory mechanisms can lead to: ① Excessively rapid heart rate, even tachyarrhythmias. This not only increases myocardial oxygen consumption but also affects ventricular filling during diastole and coronary perfusion; ② Excessive contraction of small stirred pulses, causing abnormally increased peripheral vascular resistance and elevated cardiac afterload. Moreover, the non-selective enhancement of peripheral vasoconstriction by renin, angiotensin, and vasopressin can also impair blood supply to vital organs. At this stage, vasoconstrictive responses dominate, far outweighing the vasodilatory effects of atrial natriuretic peptide and prostaglandins E2 and I2; ③ Water and sodium retention, increased blood volume, along with venous contraction, significantly elevate cardiac preload. When left ventricular filling pressure exceeds 2.0–2.4 kPa (15–18 mmHg), stroke volume no longer increases or even decreases, leading to obvious circulatory congestion and peripheral edema. Ultimately, this results in heart failure characterized by low cardiac output, high peripheral resistance, and circulatory congestion.
bubble_chart Clinical Manifestations
The main clinical manifestation of congestive heart failure is "congestion," followed by inadequate perfusion of peripheral tissues. Clinically, it is customary to classify heart failure based on which side it initially occurs and the primary site of congestion, dividing it into left-sided heart failure, right-sided heart failure, and total heart failure. Heart failure that begins in the left side of the heart and is primarily characterized by pulmonary congestion is called left-sided heart failure; heart failure that begins in the right side of the heart and is primarily characterized by congestion in organs such as the liver and kidneys, as well as peripheral venous stasis, is called right-sided heart failure. When both are present simultaneously, it is termed total heart failure. Left-sided heart failure is more common initially, often progressing over time to pulmonary hypertension and leading to right-sided heart failure. Isolated right-sided heart failure is relatively rare.
Left-sided heart failure
can be divided into two types: left ventricular failure and left atrial failure. Left ventricular failure is commonly seen in hypertensive heart disease, coronary artery disease, aortic valve disorders, and mitral regurgitation. Acute glomerulonephritis and rheumatic carditis are common disease causes of left ventricular failure in children and adolescents. In mitral stenosis, the left atrial pressure is significantly elevated, and pulmonary congestion is also present, but this is not caused by left ventricular failure and is thus referred to as left atrial failure.
(1) Symptoms
1. Dyspnea is the most prominent symptom of left-sided heart failure. In pulmonary congestion, lung tissue edema increases airway resistance and reduces alveolar elasticity, causing the alveolar wall tension to rise to the level that triggers reflexive exhalation with even small amounts of inhaled gas. This results in dyspnea, which is shallow and rapid. The degree of pulmonary congestion varies under different circumstances, leading to different forms of dyspnea.
(1) Exertional dyspnea: Initially, it occurs only after strenuous activity or physical exertion, such as climbing stairs, walking uphill, or brisk walking on level ground. As pulmonary congestion worsens, dyspnea may develop with lighter activities or even at rest.
(2) Orthopnea: A condition in which severe dyspnea while lying flat necessitates the use of high pillows, semi-recumbent positioning, or sitting upright to relieve or alleviate the dyspnea. In milder cases, dyspnea is absent when using high pillows or in a semi-recumbent position; in severe cases, sitting upright is required; in the most extreme cases, even sitting upright at the bedside with legs dangling, leaning forward, and gripping the bedside tightly may not relieve the severe dyspnea.
(3) Paroxysmal nocturnal dyspnea: Also known as cardiac asthma, it is a classic early manifestation of left ventricular failure. Dyspnea may occur consecutively for several nights, either nightly or intermittently. Typical episodes often occur 1–2 hours after falling asleep at night, when the patient suddenly awakens due to breathlessness and gasping, forced to sit up immediately, possibly accompanied by paroxysmal coughing, wheezing, or frothy sputum. In milder cases, dyspnea subsides within ten minutes to an hour after sitting up, allowing the patient to lie down and sleep again, with no noticeable symptoms during the day. In severe cases, the episode may persist, with bouts of coughing and pink frothy sputum, potentially progressing to acute pulmonary edema. Because early dyspnea often occurs at night and may resolve spontaneously, daytime symptoms may be minimal, leading the patient to overlook it. Even when seeking medical attention, the absence of positive signs of heart failure may lead to its neglect. Episodes accompanied by paroxysmal coughing or wheezing may be misdiagnosed as bronchitis or asthma.
The mechanism of paroxysmal nocturnal dyspnea is similar to that of orthopnea, possibly related to the fact that more lung tissue is below heart level when lying down, exacerbating pulmonary congestion. Additionally, the redistribution of peripheral edema fluid while lying down increases blood volume, further burdening the heart.
(4) Acute pulmonary edema: The manifestations of acute pulmonary edema are the same as those of acute left ventricular dysfunction.
2. Fatigue and lack of strength may be manifestations of low cardiac output.
3. Cheyne-Stokes respiration Seen in severe heart failure, indicating poor prognosis. Breathing rhythmically starts from a pause, gradually accelerates and deepens, then gradually slows down and becomes shallow until it stops again. After about half to one minute, breathing resumes, repeating this cycle. The mechanism is that during heart failure, cerebral ischemia and hypoxia reduce the sensitivity of the respiratory center, weakening respiration. Only when carbon dioxide accumulates to a certain level can it stimulate the respiratory center, causing breathing to accelerate and deepen. As carbon dioxide is expelled, the respiratory center gradually returns to an inhibited state, and breathing weakens until it pauses again. Patients with severe cerebral hypoxia may also experience mental symptoms such as drowsiness, dysphoria, and confusion.
(II) Signs
1. Signs of pre-existing heart disease.
2. Left ventricular enlargement The apex beat shifts downward and to the left, heart rate increases, a diastolic gallop rhythm is heard at the apex, and the second heart sound is accentuated at the pulmonary valve area. Among these, the diastolic gallop rhythm holds the most diagnostic value and is more easily heard when the patient's heart rate increases or when lying on the left side with deep exhalation. Left ventricular enlargement may also lead to relative mitral insufficiency, producing a systolic murmur at the apex.
3. Pulsus alternans Alternating strong and weak pulses. Grade I pulsus alternans can only be detected during blood pressure measurement.
4. Pulmonary rales Although some patients with left-sided heart failure may not exhibit pulmonary rales during the interstitial edema stage, and pulmonary congestion can only be detected via X-ray examination, fine moist rales at the bases of both lungs are still considered one of the important signs of left-sided heart failure. Coarse moist rales may be present during paroxysmal dyspnea or acute pulmonary edema, spreading across both lungs and possibly accompanied by wheezing.
5. Pleural effusion About 25% of patients with left-sided heart failure develop pleural effusion. The effusion may be confined to the interlobar fissures or appear as unilateral or bilateral pleural effusion, with high protein content, which subsides as heart failure improves.
(III) Early X-ray findings
During the pulmonary venous engorgement stage of left-sided heart failure, X-ray examination reveals only dilation of the upper lobe veins and relatively thinner lower lobe veins, with clear hilar vascular shadows. In the interstitial edema stage, the hilar vascular shadows appear thickened and blurred, with dilated and thickened pulmonary vascular branches or interlobar lymphatic dilation. In the alveolar edema stage, initially, high-density millet-like granular shadows are seen, which later develop into hazy cloud-like shadows. In acute pulmonary edema, fan-shaped hazy shadows extending from the hilum to the middle and peripheral lung fields are observed. Additionally, localized interlobar, unilateral, or bilateral pleural effusion may be seen in left-sided heart failure; chronic left-sided heart failure may also present with thickened interlobar pleura, and the cardiac shadow may enlarge (left ventricular enlargement).
Right-sided heart failure
Most often arises from left-sided heart failure. Once right-sided heart failure develops, due to reduced right ventricular output, pulmonary congestion often lessens, and dyspnea subsequently improves. Isolated right-sided heart failure is usually caused by acute or chronic pulmonary heart disease.
(I) Symptoms Mainly caused by chronic persistent congestion leading to functional changes in various organs, such as long-term gastrointestinal congestion resulting in loss of appetite, nausea, vomiting, etc.; renal congestion causing reduced urine output, nocturia, proteinuria, and decreased renal function; hepatic congestion leading to upper abdominal fullness or even severe abdominal pain, with long-term hepatic congestion potentially causing jaundice and cardiogenic cirrhosis.
(II) Signs
1. Signs of pre-existing heart disease.
2. Cardiac enlargement Predominantly right ventricular enlargement may be accompanied by a heaving precordial impulse (forceful and sustained cardiac pulsation at the left sternal border). Heart rate increases, and some patients may exhibit an early diastolic gallop rhythm heard over the right ventricular surface at the left sternal border. Significant right ventricular dilation may lead to functional tricuspid insufficiency, producing a systolic murmur at the tricuspid area that intensifies during inspiration.
3. Venous engorgement Engorgement of the external jugular vein is an early sign of right-sided heart failure. When in a semi-recumbent or seated position, engorgement of the external jugular vein above the clavicle or a highest engorgement point more than 10 cm above the sternal angle indicates elevated venous pressure, often more pronounced on the right side. In severe right-sided heart failure with markedly elevated venous pressure, dorsal hand veins and other superficial veins may also become engorged, with visible venous pulsations.
4. Hepatomegaly and tenderness appear relatively early, often occurring before subcutaneous edema. The liver enlargement is more pronounced below the xiphoid process than at the costal margin, with a softer texture and a sense of fullness. The edge may sometimes be indistinct on palpation, and dullness is noted on percussion below the xiphoid process, accompanied by tenderness. Compression of the liver (or the dull area below the xiphoid process) may exacerbate jugular vein distension (hepatojugular reflux phenomenon). The degree of hepatomegaly may decrease or increase rapidly in response to improvements or worsening of heart failure. When right heart failure suddenly worsens, acute hepatic congestion and central lobular necrosis can lead to a rapid increase in liver size, accompanied by severe pain in the right upper quadrant and below the xiphoid process, along with significant tenderness and jaundice. Concurrently, serum alanine aminotransferase (ALT) levels often rise markedly, sometimes exceeding 1000U in some cases. Once heart failure improves, hepatomegaly and jaundice subside, and serum transaminase levels typically return to normal within 1–2 weeks. In cases of long-term chronic right-sided heart failure leading to cardiac cirrhosis, the liver feels firmer on palpation, tenderness may be less pronounced, and it is often accompanied by jaundice, ascites, and chronic liver dysfunction.
5. Dependent edema In the early stages of right-sided heart failure, edema is often not obvious and usually appears after jugular vein distension and hepatomegaly become more pronounced. Initially, there is subcutaneous fluid accumulation and weight gain, and only after reaching a certain degree does pitting edema develop. Edema first appears in the dependent parts of the body. In ambulatory patients, it is more noticeable in the feet, medial ankles, and pretibial areas; in those lying supine, sacral edema occurs; and in those lying on their side, edema is more prominent in the dependent limb. In severe cases, generalized edema may develop.
The mechanism of edema formation has not yet been fully elucidated. Numerous studies have shown that reduced renal sodium excretion is a result of renal blood flow redistribution—under conditions of decreased total renal blood flow, perfusion to the outer renal cortex decreases, while perfusion to the inner cortex and outer medulla increases relatively. Nephrons in the inner cortex and outer medulla have longer loops of Henle and a stronger capacity for sodium reabsorption. The relative increase in blood flow to these areas enhances sodium reabsorption and retention. Additionally, some suggest that when renal blood flow decreases, both the glomerular filtration rate (GFR) and renal plasma flow decline, but the GFR decreases less than renal plasma flow, resulting in a relatively higher filtration fraction. This increases the plasma protein content in the peritubular capillaries, raising the colloid osmotic pressure and promoting increased proximal tubule reabsorption, leading to sodium and water retention. Other factors, such as increased renal venous pressure, reduced GFR, and excessive secretion or decreased inactivation of aldosterone and antidiuretic hormone, though not primary, may also contribute to the pathogenesis.
6. Pleural effusion and ascites The pleural membrane veins drain into the superior vena cava, bronchial veins, and pulmonary veins. With right-sided heart failure, increased venous pressure can lead to bilateral or unilateral pleural effusion. In bilateral effusions, the right side is often more significant; unilateral effusions also tend to occur more frequently on the right side, though the reason is unclear. The pleural fluid has a high protein content (approximately 2–3 g/100 ml) with normal cell counts. Significant ascites is commonly seen in tricuspid stenosis, Ebstein's anomaly, and constrictive pericarditis, as well as in advanced-stage heart failure and when a right atrial thrombus obstructs the inferior vena cava.
7. Pericardial effusion Small amounts of pericardial effusion are not uncommon in right-sided or total heart failure. It is often detected via echocardiography or autopsy and does not cause cardiac tamponade symptoms.
8. Cyanosis Most patients with long-standing right-sided heart failure exhibit cyanosis, manifesting as facial capillary dilation, cyanosis, and pigmentation. Cyanosis results from increased tissue oxygen extraction due to inadequate blood supply, leading to low venous oxygen levels.
9. Advanced-stage patients may show significant malnutrition, weight loss, or even cachexia.
(三)Laboratory findings
1. Increased venous pressure A cubital venous pressure exceeding 1.4 kPa (14 cmH2O) or a rise of 0.1–0.2 kPa (1–2 cmH2O) after firm liver pressure for ½–1 minute suggests right-sided heart failure (normal range in 1,425 Chinese adults: 0.3–1.4 kPa (3–14 cmH2O), average 0.99 kPa (9.9 cmH2O)).
2. Slight increases in serum bilirubin and alanine aminotransferase may occur.
3. Urinary changes Grade I proteinuria, small numbers of hyaline or granular casts, and occasional red blood cells may be present, along with grade I azotemia.
(四)X-ray findings These may reveal cardiomegaly, widened superior vena cava, enlarged right atrium and ventricle, and possible bilateral or unilateral pleural effusion.
Diastolic heart failure
The pumping function of the heart relies on ventricular contraction to expel blood and rapid refilling during subsequent relaxation. Diastolic heart failure refers to a condition where ventricular systolic function is normal, but rapid refilling is impaired, leading to reduced ventricular filling volume and/or increased filling pressure, decreased stroke volume, and consequently heart failure. It is mostly caused by ventricular wall hypertrophy and/or increased stiffness, with little or no ventricular enlargement. The main disease causes include long-term hypertension, hypertrophic cardiomyopathy, left ventricular outflow tract obstruction, coronary artery disease, and restrictive cardiomyopathy. Aging, diabetes, acute right ventricular enlargement restricting left ventricular filling, and pericardial diseases also affect ventricular relaxation. Whether these factors should be included as disease causes of diastolic heart failure remains controversial. Some disease causes of diastolic heart failure may also impair ventricular systolic function, but diastolic dysfunction may occur earlier.
The main clinical manifestation of diastolic heart failure is pulmonary static blood. In the early stages, compensation may occur through increased atrial pressure and/or enhanced atrial contraction, making pulmonary static blood symptoms less apparent. However, ventricular filling often fails to accelerate correspondingly during exercise, leading to varying degrees of reduced exercise tolerance. In severe cases, varying degrees of dyspnea or even pulmonary edema may occur. When heart rate increases or supraventricular tachyarrhythmias such as atrial fibrillation occur, pulmonary static blood manifestations worsen. Low stroke volume during exercise can lead to syncope.
X-ray examination: The cardiac shadow is mostly not enlarged, but varying degrees of pulmonary congestion may be observed.
Echocardiography: Currently, Doppler echocardiography is mostly used to indirectly measure ventricular diastolic function via the mitral valve blood flow spectrum. Observation indicators include isovolumic relaxation time (IVRT), deceleration time of early diastolic filling (DT), early and advanced stage filling velocities and their ratio (E, A, and E/A). Slowed left ventricular myocardial relaxation is manifested by a low E peak, high A peak, decreased E/A, and prolonged IVRT; increased left ventricular myocardial stiffness results in a high E peak, low A peak, increased E/A, and shortened IVRT. When reduced left ventricular relaxation is combined with increased myocardial stiffness, the combined effects of these changes can cause the mitral valve spectrum to appear "pseudo-normalized." In such cases, it is necessary to simultaneously measure the atrial systolic pulmonary venous flow reversal velocity (AR) and the time interval from the onset of atrial contraction to the onset of pre-systolic velocity in the left ventricular outflow tract (Ar). Elevated AR (>20 cm/s) and shortened A-Ar (<45 ms) indicate abnormal ventricular diastolic function. Normal reference values for IVRT, E, A, and E/A in young and elderly individuals are shown in Table 7.
Table 7 Normal reference values for common diastolic function indicators in young and elderly individuals
Group | IVRT (ms) | E (cm/s) | A (cm/s) | E/A |
Young individuals | 72±12 | 69±12 | 27±7 | 2.7±0.7 |
Elderly individuals | 84±12 | 59±14 | 46±13 | 1.2±0.4 |
Asymptomatic heart failure
Asymptomatic heart failure, also known as asymptomatic left ventricular dysfunction, has been extensively studied, particularly in cases of asymptomatic left ventricular systolic dysfunction following myocardial infarction. Data on asymptomatic left ventricular diastolic dysfunction is scarce. Asymptomatic left ventricular dysfunction is defined as the absence of typical congestive heart failure symptoms, without the need for digitalis or diuretic treatment, but with objective evidence of left ventricular dysfunction (e.g., left ventricular ejection fraction [LVEF] below 40% or X-ray showing pulmonary blood flow redistribution indicating grade I pulmonary congestion), with cardiac function classified as NYHA class I. Asymptomatic left ventricular systolic dysfunction represents the preclinical stage of symptomatic heart failure. During this phase, the condition appears relatively stable on the surface, but internal myocardial remodeling and ventricular reshaping continue to progress. The adaptive state will eventually transition into maladaptation, leading to the development of symptomatic heart failure. The duration of the asymptomatic heart failure stage varies widely, ranging from weeks to years, influenced by factors such as the patient's age, heart size, LVEF value, the initial cause of myocardial damage, the progression of underlying diseases, and genetic factors. Clinical studies have confirmed that in placebo-controlled groups of patients with asymptomatic left ventricular dysfunction years after myocardial infarction, the left ventricle continues to dilate, albeit at a slower rate compared to symptomatic heart failure patients. However, during long-term follow-up, the incidence of symptomatic heart failure and mortality rates due to heart failure remain high. ACE inhibitor therapy significantly reduces and delays the onset of symptomatic heart failure. Common objective indicators for left ventricular systolic dysfunction include LVEF (measured by Doppler echocardiography or radionuclide scanning), as well as left ventricular fractional shortening (by echocardiography) and assessment of left ventricular wall motion abnormalities. An LVEF below 40% or a left ventricular fractional shortening rate below 1.1 circumferences per second suggests left ventricular systolic dysfunction. Left ventricular wall systolic bulging, akinesis, hypokinesis, or dyskinesis are all considered wall motion abnormalities, and their evaluation should take into account the severity, extent of the abnormalities, and LVEF.
Assessment and Classification of Cardiac Function
Cardiac function refers to the limit of the heart's working capacity.
(1) NYHA Functional Classification: A classification by the New York Heart Association based on patients' subjective symptoms. Class I: No limitation of physical activity; ordinary physical activity does not cause excessive lack of strength, palpitation, shortness of breath, or angina. Class II: Slight limitation of physical activity; comfortable at rest, but less than ordinary activity causes lack of strength, palpitation, shortness of breath, or angina. Class III: Marked limitation of physical activity; comfortable at rest, but less than ordinary activity causes lack of strength, palpitation, shortness of breath, or angina. Class IV: Unable to carry out any physical activity without discomfort; symptoms of heart failure or angina may be present at rest, and any physical activity increases discomfort.
In March 1994, the above classification was revised to include objective assessment indicators (such as ECG, stress tests, X-rays, echocardiography, and nuclear imaging results) as follows: A. No objective evidence of cardiovascular disease. B. Objective evidence of mild cardiovascular disease. C. Objective evidence of moderate cardiovascular disease. D. Objective evidence of severe cardiovascular disease. The definitions of mild, moderate, and severe cardiovascular disease are difficult to specify precisely and are subject to the clinician's judgment.
Combining symptoms and objective indicators may compensate for the shortcomings of the original scheme, which separated subjective symptoms from objective indicators and only reflected hemodynamic symptom changes. For example, a patient with severe aortic stenosis or severe coronary artery stenosis but minimal or no symptoms would be classified as ID. Conversely, an asymptomatic patient with mild aortic stenosis or mild coronary artery stenosis would be classified as IB. Similarly, asymptomatic patients with LVEF <35% would be classified as IC, while those with symptomatic heart failure would be classified as II–IVC.
This classification is simple and practical. The newly revised combined indicator classification is very helpful in comparing the cardiac functional status of subjects in different clinical trials, evaluating treatment effects, and analyzing the impact of treatment on different subgroups.
(2) Exercise Tolerance Testing and Classification: Graded exercise tests are commonly performed using a treadmill or bicycle ergometer, with symptom-limited maximal or heart rate-limited submaximal intensity as the endpoint. ECG, nuclear cardiovascular angiography, echocardiography, or continuous measurement of end-tidal O2 and CO2 concentrations are used to assess the patient's response to exercise. Observation indicators include total exercise time, exercise workload (measured in metabolic equivalents, METs; 1 MET is the oxygen consumption at rest, equivalent to 3.5 ml·min-1/kg), the degree of increase in left ventricular ejection fraction during exercise, maximal oxygen uptake (VO2max), and anaerobic threshold (AT). Total exercise time must be converted to METs based on the exercise protocol to reflect exercise tolerance, but when the same protocol is used, it can compare exercise tolerance before and after treatment. The evaluation criteria are normal if the exercise workload is 6–10 METs, LVEF increases by >5% during exercise, maximal oxygen uptake is >20 ml·min-1/kg, and anaerobic threshold is >14 ml·min-1/kg. Based on the degree of reduction in VO2max and AT, Weber classified cardiac function into four grades: A, B, C, and D (Table 8).
Table 8 Weber’s Exercise Tolerance (VO2max and AT) Classification
| Classification | Degree of Cardiac Function Impairment | VO2max (ml·min-1/kg) | AT (ml·min-1/kg) | Peak CI (L·min-1/m²) |
| A | None→Grade I | >20 | >14 | 8 |
| B | Mild→Grade II | 16–20 | 11–14 | 6–8 |
| C | Moderate→Grade III | 10–16 | 8–11 | 4–6 |
| D | Grade III | <10 | <8 | <4 |
[Note] VO2max: Maximum oxygen uptake, the point where oxygen uptake plateaus for ≥30s despite continued exercise. AT: Anaerobic threshold, the point where oxygen uptake and carbon dioxide output become disproportionate, equivalent to ~70% of VO2max level.
Due to varying responses to the same treatment among heart failure patients with different disease etiologies, progression stages, and clinical manifestations, modern heart failure therapy increasingly emphasizes selecting optimal individualized treatment plans tailored to specific subgroups and cardiac function states. The assessment of systolic or diastolic dysfunction and cardiac function status holds significant reference value for determining therapeutic strategies.
The diagnosis of typical heart failure is not difficult. The diagnosis of left-sided heart failure is based on the signs of pre-existing heart disease and manifestations of pulmonary congestion. The diagnosis of right-sided heart failure is based on the signs of pre-existing heart disease and manifestations of systemic static blood, and most patients have a history of left-sided heart failure.
It is noteworthy to emphasize the early diagnosis of heart failure. Patients with early-stage heart failure may have subtle symptoms, often able to move freely and continue working. Exertional dyspnea and paroxysmal nocturnal dyspnea are early symptoms of left-sided heart failure, but they often go unnoticed and may be overlooked due to the absence of positive signs during daytime consultations. If a detailed medical history is not taken, a thorough examination is not performed, and typical findings such as diastolic gallop rhythm or characteristic X-ray manifestations are missed, it can easily be misdiagnosed as fistula disease. Jugular vein distension and hepatomegaly are early signs of right-sided heart failure but are often overlooked. For instance, general physical examinations may not pay attention to the jugular veins, and in heart failure, hepatomegaly is often located below the xiphoid process, not palpable below the costal margin. Even if hepatomegaly is detected, it may not be attributed to heart failure if not accompanied by dyspnea or edema, and tests like hepatojugular reflux are neglected. Some symptoms and signs of heart failure can also occur in other diseases. Therefore, dyspnea, edema, and hepatomegaly in patients with heart disease are not necessarily caused by heart failure. For example, exertional dyspnea may result from obstructive lung qi swelling, pulmonary insufficiency, obesity, or general weakness. Nocturnal dyspnea can also be caused by bronchial asthma attacks. Basal lung crackles may arise from chronic bronchitis, bronchiectasis, or pneumonia. Crackles due to heart failure are usually bilateral and symmetrical, though occasionally unilateral or presenting solely as wheezing. Lower limb edema can be caused by varicose veins, phlebitis, kidney or liver diseases, lymphedema, or may occur after prolonged sitting, during menstruation, or in the late stages of pregnancy [third trimester]. Idiopathic lower limb edema in women is also not uncommon. Additionally, in heart failure, prolonged bed rest may lead to fluid accumulation in the lumbosacral region rather than lower limb edema. Hepatomegaly can result from schistosomiasis, hepatitis, or fatty liver. Jugular vein distension may be due to lung qi swelling or mediastinal tumors compressing the superior vena cava. Pleural effusion can be caused by pleural membrane subcutaneous nodules, tumors, or infarction; ascites may also arise from cirrhosis, hypoalbuminemia, peritoneal membrane subcutaneous nodules, or tumors.
Heart failure is often accompanied by cardiac enlargement, but it can also occur in a normal-sized heart, such as in acute myocardial infarction. Cardiac enlargement may be masked by lung qi swelling, while cardiac displacement or pericardial effusion can be mistaken for cardiac enlargement. Thus, to accurately diagnose heart failure and avoid misdiagnosis or fistula disease diagnosis, it is essential to take a detailed medical history, conduct a thorough examination, and perform a comprehensive analysis combining the symptoms and signs of heart disease and heart failure.
bubble_chart Treatment Measures
In recent years, significant progress has been made in the prevention and treatment of systolic heart failure. Methods for evaluating efficacy have expanded beyond improvements in symptoms, hemodynamic effects, exercise tolerance, and quality of life to include long-term treatment safety, mortality rates, survival duration, and the degree of neurohormonal system activation. Prevention strategies increasingly emphasize the importance of halting the formation and progression of heart failure. For asymptomatic and Grade I symptomatic heart failure, angiotensin-converting enzyme inhibitors (ACEIs) are recommended to improve prognosis. For Grade III symptomatic heart failure, ACEIs combined with diuretics and/or digoxin are also advisable to alleviate symptoms, reduce disability, and prolong survival. Specific prevention and treatment measures include:
(1) Prevention and treatment of disease causes
Rheumatic valvular heart disease remains a common cause of chronic heart failure in China. The use of penicillin to treat streptococcal infections has nearly eradicated rheumatic fever and rheumatic valvular heart disease in developed countries. Elective surgical treatment for rheumatic valvular heart disease, effective control of hypertension, and active prevention and treatment of coronary artery disease and myocardial ischemia, among other disease causes, along with eliminating triggers of heart failure such as infections, excessive physical exertion, and mental stress, can prevent the onset of heart failure.
(2) Treatment of systolic heart failure
1. Reducing cardiac load This includes minimizing physical activity and mental stress. Severe cases should adhere to strict bed rest, with gradual increases in activity as cardiac function improves to avoid complications such as venous thrombosis or pneumonia from prolonged bed rest. Additionally, addressing psychological burdens is important, and small doses of sedatives may be administered if necessary.
2. Restricting sodium intake Daily dietary sodium intake should be appropriately limited to 2–5 grams of salt, avoiding salted or preserved foods. When diuretics cause significant diuresis, sodium restriction should not be overly strict to prevent hyponatremia.
3. Use of diuretics Diuretics inhibit sodium (Na+) reabsorption at different sites of the renal tubules or increase glomerular Na+ filtration, promoting the excretion of water and Na+, thereby reducing ventricular filling pressure and alleviating clinical symptoms caused by pulmonary and/or systemic circulatory congestion. While their efficacy is well-established, their impact on the overall course of heart failure (e.g., survival rates) remains unclear. Long-term use of diuretics may theoretically lead to adverse effects such as: ① Reduced cardiac output, activating the renin-angiotensin system (RAS) and increasing plasma renin and aldosterone levels. ② Hypokalemia. ③ Impaired glucose tolerance. ④ Hyperuricemia. ⑤ Hyperlipidemia. ⑥ Ventricular arrhythmias. Currently, diuretics are first-line drugs for treating heart failure with fluid and sodium retention, often used in combination with other heart failure medications (e.g., digoxin, ACE inhibitors). They should be used cautiously in cases of pure diastolic heart failure.
Commonly used diuretics include:
(1) Thiazide and thiazide-like diuretics (Table 1): These act on the distal convoluted tubule and the distal ascending limb of the loop of Henle, inhibiting Na+ reabsorption. Their diuretic effect is moderate. When the glomerular filtration rate falls below 30 ml/min, their efficacy is significantly limited, making them unsuitable for severe heart failure (with markedly reduced renal blood flow) or patients with chronic renal insufficiency. Metolazone differs from hydrochlorothiazide and similar agents in that its diuretic effect is not diminished in renal impairment. Its action extends beyond the distal tubule and ascending limb, possibly affecting the proximal tubule as well. It has a prolonged diuretic effect, with a single dose lasting 12–24 hours. When combined with furosemide, it produces an excellent diuretic response and is highly effective for patients with renal insufficiency.
Table 1: Dosages and duration of action of different thiazide and thiazide-like diuretics
Preparation Name | dose(mg/d) | Duration of Action(h) | |
Hydrochlorothiazide | hydrochlorothiazide | 12.5~50 | 1~12 |
Hydroflumethiazide | hydroflumethiazide | 25~50 | 4~6 |
Chlorthalidone | chlorthalidone | 12.5~50 | 24~72 |
Metolazone | metolazone | 1~10 | 18~25 |
Chlorothiazide | chlorothiazide | 250~1000 | - |
Cyclopenthiazide | cyclopenthiazide | 0.25 | - |
(2) Loop diuretics (Table 2): They act on the thick ascending limb of the loop of Henle, inhibiting the reabsorption of Cl- and Na+ at this site. This results in a high concentration of Na+ in the urine reaching the distal tubule, leading to the excretion of large amounts of Na+ and water, thereby producing a potent


